
Fall Project 2: Das Lab
One of the most sought after applications necessary for de novo design of riboswitches is to accurately predict riboswitch binding affinities to their ligands. Before the revolution of transformer-based LLMs for macromolecule structure prediction, EternaBench was created as a diverse riboswitch benchmark dataset for training and evaluation of physics-based and probabilistic ML models, including EternaFold. The designed riboswitches in this experimental dataset each included two aptamers: the first an aptamer for an input ligand and the second for an output reporter fluorescently tagged MS2 viral coat protein. These riboswitches were designed so that upon binding of the input ligand (flavin mononucleotide (FMN), theophylline, or tryptophan), the MS2 protein aptamer is more accessible, thereby increasing binding affinity of fluorescent protein. Measurements were performed on this diverse dataset for MS2 protein binding affinities with and without ligand present (K+lig and K-lig).
Previously, I indirectly evaluated the potential for selected probabilistic, thermodynamic, and deep learning models at predicting ligand-binding affinity through a zero-shot approach by extracting the probability of the terminal base pair in the hairpin of the MS2 protein aptamer.
However, I furthermore wanted to fine-tune RibonanzaNet to predict ligand-binding affinity, and then evaluate predictions against models of which a constrained partition function calculation was used to predict ligand binding affinities. All riboswitches have 1 of three ligand-binding aptamers with theoretically the same binding affinity. However, the prediction task for models is to predict the binding affinity of the MS2 protein aptamer to the MS2 protein, both with and without ligand present. Here is a table of the binding affinities for each ligand :
| Ligand | Aptamer sequence | Aptamer constraint | Concentration | Kd (μM) |
|---|---|---|---|---|
| FMN | nAGGAUAU&AGAAGGn | (xxxxxx(&)xxxxx) | 200 μM | 2.2 |
| Tryptophan | AGGACCGG&CCGCCACU | ((xxx(((&)))xxx)) | 2.4 mM | 1.3 |
| Theophylline | GAUACCAG&CCCUUGGCAGC | (xxx((((&)xxx)))xxx) | 2.0 mM | 20 |
This table is adapted from Supplementary Table 14 from the paper RNA secondary structure packages evaluated and improved by high-throughput experiments .
My first attempt at fine-tuning involved manipulating how the model processed the sequence features. The most simple example inlcuded just passing the mean of the sequence feature into a linear layer with an output of 2 (for both the log of the riboswitch KD with and without ligand bound). However, my attempts at processing the sequence features did not work, and predictions on the test set were essentially the same for each sequence. This makes sense because averaging the sequence embeddings effectively collapses all positional and relational information. Since the global RNA structure is the key component to predicting ligand binding and recognition, during fine-tuning the model was unable to learn anything meaningful.
My second attempt at fine-tuning involved manipulating the pairwise features . My inital simple manipulation techinques did not work (including just averaging the flattened pairwise features before passing through a linear layer). This makes sense for the same reason averaging the sequence embeddings directly did not work. However, when I averaged each structural channel of the pairwise features, annd then averaged the pooled channels before passing it through a linear layer, I allowed the model to pick up on the relationship between ligand binding and RNA secondary/tertiary structure.
I actually fine-tuned RibonanzaNet on this data (which I called the fine-tuned model “RibonanzaNet-EB” ) within a Jupyter NB with a gpu backend through a slurm supercomputer. You can see the Training NBs Here. You can download the training data from the archived EB repository and RibonanzaNet. You can also use another RNA-LLM .
Now using the same training /test split as EternaFold, here are some results :
Comparing PCC with EternaFold :
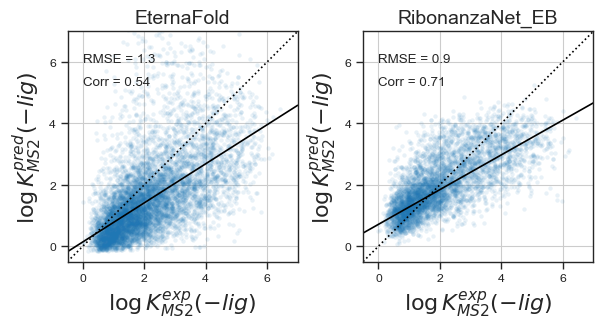
Because many if not most riboswitches in the EternaBench dataset did not have significant differences in MS2 binding affinity with and without ligand bound, to test whether the model can predict RNA structure with ligand bound I also fine-tuned RibonanzaNet on the activation ratio, and evaluated results :
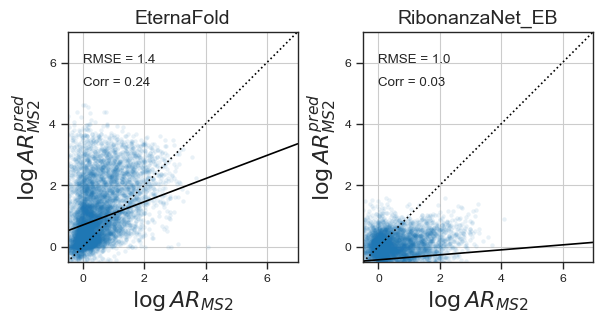
RibonanzaNet is not able to predict RNA binding affinity, given a shift in ensemble due to ligand binding. However, combined with a pretrained model on reactivity data with ligand bound (see here), I am hopeful that this would be possible.